
泛基因组 | 分享一套“数据下载、质控、组装、矫正、注释到泛基因组统计与绘图“的泛基因组分析组装代码
生信服务器活动
网址:
https://dayu.xiyoucloud.net/dayu/api/v1/anonymous/affiliate/BioinfoDu
写在前面
我们前段时间,分享了Linux中部署及使用Codex教程,若是在本地部署,也非常容易。我们直接使用Codex的API密匙即可完成部署。
对于Codex的理解和编程能力,可能未使用的人对此还是有比较大的疑惑。me too。
本期,分享一套基于Codex协助完成的泛基因组分析流程代码。
注:本套代码仅供参考学习。
若要获得本期教程数据和代码,在后台回复:20260624
结果如下
1. 项目结构
pangenome_tutorial/
├── README.md
├── docs/
│ └── pangenome_workflow.md
├── env/
│ └── pangenome_env.yml
├── script/
│ ├── 00_create_env.sh
│ ├── 01_download_public_data.sh
│ ├── 02_genome_qc_from_public_assemblies.sh
│ ├── 02_qc_trim_assemble.sh
│ ├── 03_polish_and_correct.sh
│ ├── 04_annotate.sh
│ ├── 05_run_pangenome.sh
│ ├── run_r_analysis.sh
│ └── run_r_analysis_from_existing_matrix.sh
├── R/
│ ├── 01_generate_demo_data.R
│ ├── 02_pangenome_analysis.R
│ ├── 03_plot_figures.R
│ └── 04_import_roary_panaroo.R
├── data/
│ ├── metadata/
│ └── pangenome/
└── results/
├── figures/
└── tables/
2. 研究对象与公开数据来源
1.1 参考文献
Tettelin 等 2005 年发表的 S. agalactiae 研究对多个致病菌株进行比较基因组分析,指出单个参考基因组不能代表物种内全部遗传多样性,并提出 pan-genome 的思想。文章摘要中报告,该物种的核心基因组约占单个基因组的 80%,其余为不同菌株间差异明显的附属基因组。
本教程的数据表位于:
data/metadata/tettelin_2005_accessions.tsv
data/metadata/publication.tsv
tettelin_2005_accessions.tsv 中列出了用于示例重分析的 8 个基因组入口:
| 菌株 | accession | 类型 | 说明 |
|---|---|---|---|
| 18RS21 | AAJO01000000 | WGS master | Tettelin 2005 新沉积 |
| 515 | AAJP01000000 | WGS master | Tettelin 2005 新沉积 |
| CJB111 | AAJQ01000000 | WGS master | Tettelin 2005 新沉积 |
| COH1 | AAJR01000000 | WGS master | Tettelin 2005 新沉积 |
| H36B | AAJS01000000 | WGS master | Tettelin 2005 新沉积 |
| A909 | CP000114 | complete chromosome | Tettelin 2005 新沉积 |
| NEM316 | AL732656 | complete chromosome | 已公开数据库基因组 |
| 2603V/R | AE009948 | complete chromosome | 已公开数据库基因组 |
1.2 两种可运行入口
由于该经典文章公开沉积的是组装后的 WGS/染色体序列,而不是现代 Illumina FASTQ 原始读段,本教程将流程分为两条入口。
入口 A:复现文献公开基因组数据
cd pangenome_tutorial
bash script/01_download_public_data.sh
bash script/02_genome_qc_from_public_assemblies.sh
入口 B:新项目或 SRA FASTQ 原始读段
cd pangenome_tutorial
cp data/metadata/samples_template.tsv data/metadata/samples.tsv
# 编辑 data/metadata/samples.tsv,使 fq1/fq2 指向真实 fastq.gz 文件
bash script/02_qc_trim_assemble.sh
bash script/03_polish_and_correct.sh
bash script/04_annotate.sh
bash script/05_run_pangenome.sh
2. 软件环境构建
所有软件安装定义在 env/pangenome_env.yml 中。推荐使用 mamba,因为生物信息依赖较多。
cd pangenome_tutorial
bash script/00_create_env.sh
conda activate pangenome_env
该环境包含:
| 类型 | 软件 |
|---|---|
| 数据下载 | entrez-direct, ncbi-datasets-cli, sra-tools |
| 原始 reads 质控 | FastQC, fastp, MultiQC |
| 组装与评估 | SPAdes, QUAST, CheckM |
| 矫正 | BWA, SAMtools, Pilon |
| 注释 | Prokka, Bakta |
| 泛基因组 | Panaroo, Roary, MAFFT, FastTree |
| R 分析和绘图 | R, ggplot2, tidyverse, ape, vegan, pheatmap |
Bakta 和 CheckM 需要额外数据库。真实项目建议把数据库放在项目目录下,例如:
mkdir -p data/db
bakta_db download --output data/db/bakta
export BAKTA_DB="$(pwd)/data/db/bakta/db"
mkdir -p data/db/checkm
checkm data setRoot data/db/checkm
3. 数据下载
3.1 文献公开基因组下载
script/01_download_public_data.sh 读取 data/metadata/tettelin_2005_accessions.tsv,使用 Entrez Direct 从 NCBI nucleotide 数据库下载 FASTA 和 GFF3。
bash script/01_download_public_data.sh
输出:
data/raw/genomes/*.fna
data/raw/genomes/*.gff3
logs/downloaded_genome_stats.tsv
3.2 原始 FASTQ 项目的样本表
如果分析新测序项目或 SRA 下载后的 reads,准备如下样本表:
sample_id fq1 fq2
sample_01 data/raw/reads/sample_01_R1.fastq.gz data/raw/reads/sample_01_R2.fastq.gz
sample_02 data/raw/reads/sample_02_R1.fastq.gz data/raw/reads/sample_02_R2.fastq.gz
本项目提供模板:
cp data/metadata/samples_template.tsv data/metadata/samples.tsv
4. 质控、修剪与组装
原始 reads 质控由 script/02_qc_trim_assemble.sh 完成。该脚本对每个样本执行:
- FastQC:检查原始 reads 质量。
- fastp:去接头、低质量端修剪、过滤过短 reads。
- FastQC:检查修剪后 reads。
- SPAdes
--careful:进行细菌基因组短读长组装。 - QUAST:评估 contig 数量、N50、总长度、GC 等指标。
- MultiQC:整合质控报告。
bash script/02_qc_trim_assemble.sh
推荐初筛阈值:
| 指标 | 建议阈值 | 解释 |
|---|---|---|
| Q30 比例 | 大于 80% | 低于该值说明测序质量偏差,需要谨慎 |
| adapter 含量 | 小于 5% | 高接头比例会影响组装 |
| reads 数 | 每个基因组 50 倍以上覆盖度更稳妥 | 细菌组装通常需要足够覆盖 |
| contig N50 | 越高越好 | 与数据质量和重复序列有关 |
| contig 数 | 越少越好 | 过多 contig 可能导致泛基因组假阳性 |
| 总长度 | 接近物种预期 | 明显偏大可能污染,偏小可能缺失 |
| CheckM completeness | 大于 95% | 代表基因组较完整 |
| CheckM contamination | 小于 5% | 高污染会夸大 pan-genome |
5. 基因组矫正与过滤
组装后使用 reads 回贴到 contig,通过 Pilon 矫正小 indel、替换和局部错误。
bash script/03_polish_and_correct.sh
脚本步骤:
- BWA index:为 contig 建索引。
- BWA MEM:将修剪后 reads 比对回组装结果。
- SAMtools sort/index:整理 BAM。
- Pilon:根据 read alignment 矫正组装。
- seqkit:过滤小于 500 bp 的短 contig。
矫正后的文件位于:
results/polished/<sample>/<sample>.polished.min500.fna
质量控制要点:
- 同一项目内所有样本必须采用同一组装、过滤和矫正策略。
- 不建议把完成图基因组、草图基因组、不同测序平台结果不加说明地直接混合。
- 若泛基因组中 singleton 异常偏多,应优先检查污染、碎片化和注释一致性,而不是直接解释为生物学差异。
6. 基因组注释
泛基因组分析依赖基因预测和功能注释。不同注释器会改变 CDS 边界和基因数量,因此同一个项目必须统一注释流程。
bash script/04_annotate.sh
默认执行 Prokka,若设置 BAKTA_DB 则同时执行 Bakta。
Prokka 输出:
results/annotation/prokka/<sample>/<sample>.gff
results/annotation/prokka/<sample>/<sample>.faa
results/annotation/prokka/<sample>/<sample>.ffn
推荐实践:
- 泛基因组聚类使用同一注释器产生的 GFF。
- 发表时报告注释软件版本、数据库版本和关键参数。
- 物种名、菌株名、locustag 应统一且不包含空格。
7. 泛基因组构建
本项目同时给出 Panaroo 和 Roary。Roary 是经典工具,适合快速得到基因 presence/absence 矩阵;Panaroo 更强调错误基因、碎片基因和图结构清理,适合对草图组装进行稳健分析。
bash script/05_run_pangenome.sh
核心参数:
panaroo \
-i *.gff \
-o results/pangenome/panaroo \
--clean-mode strict \
--remove-invalid-genes \
-a core
roary \
-e --mafft \
-p 16 \
-f results/pangenome/roary \
*.gff
常见结果文件:
| 文件 | 来源 | 用途 |
|---|---|---|
gene_presence_absence.Rtab |
Panaroo/Roary | R 绘图和统计矩阵 |
gene_presence_absence.csv |
Panaroo/Roary | 含注释的宽表 |
core_gene_alignment.aln |
Panaroo/Roary | 核心基因系统发育 |
summary_statistics.txt |
Panaroo/Roary | pan/core/shell/cloud 摘要 |
8. R 语言下游分析与绘图
8.1 演示数据
本教程已生成可直接运行的演示矩阵:
data/pangenome/gene_presence_absence_demo.tsv
data/pangenome/gene_family_annotation_demo.tsv
data/pangenome/per_genome_gene_counts_demo.tsv
生成、分析和绘图:
bash script/run_r_analysis.sh
该命令依次运行:
R/01_generate_demo_data.R
R/02_pangenome_analysis.R
R/03_plot_figures.R
8.2 导入真实 Panaroo/Roary 矩阵
真实项目跑完 Panaroo 后,导入矩阵:
Rscript R/04_import_roary_panaroo.R "$(pwd)" results/pangenome/panaroo/gene_presence_absence.Rtab
bash script/run_r_analysis_from_existing_matrix.sh
如果使用 Roary 输出:
Rscript R/04_import_roary_panaroo.R "$(pwd)" results/pangenome/roary/gene_presence_absence.Rtab
bash script/run_r_analysis_from_existing_matrix.sh
8.3 R 分析内容
R/02_pangenome_analysis.R 完成以下统计:
- 计算每个 gene family 在多少个基因组中出现。
- 分类为 Core、Soft-core、Shell、Cloud。
- 统计每个样本的核心与附属基因数量。
- 通过 200 次随机基因组加入顺序估计 pan/core 累积曲线。
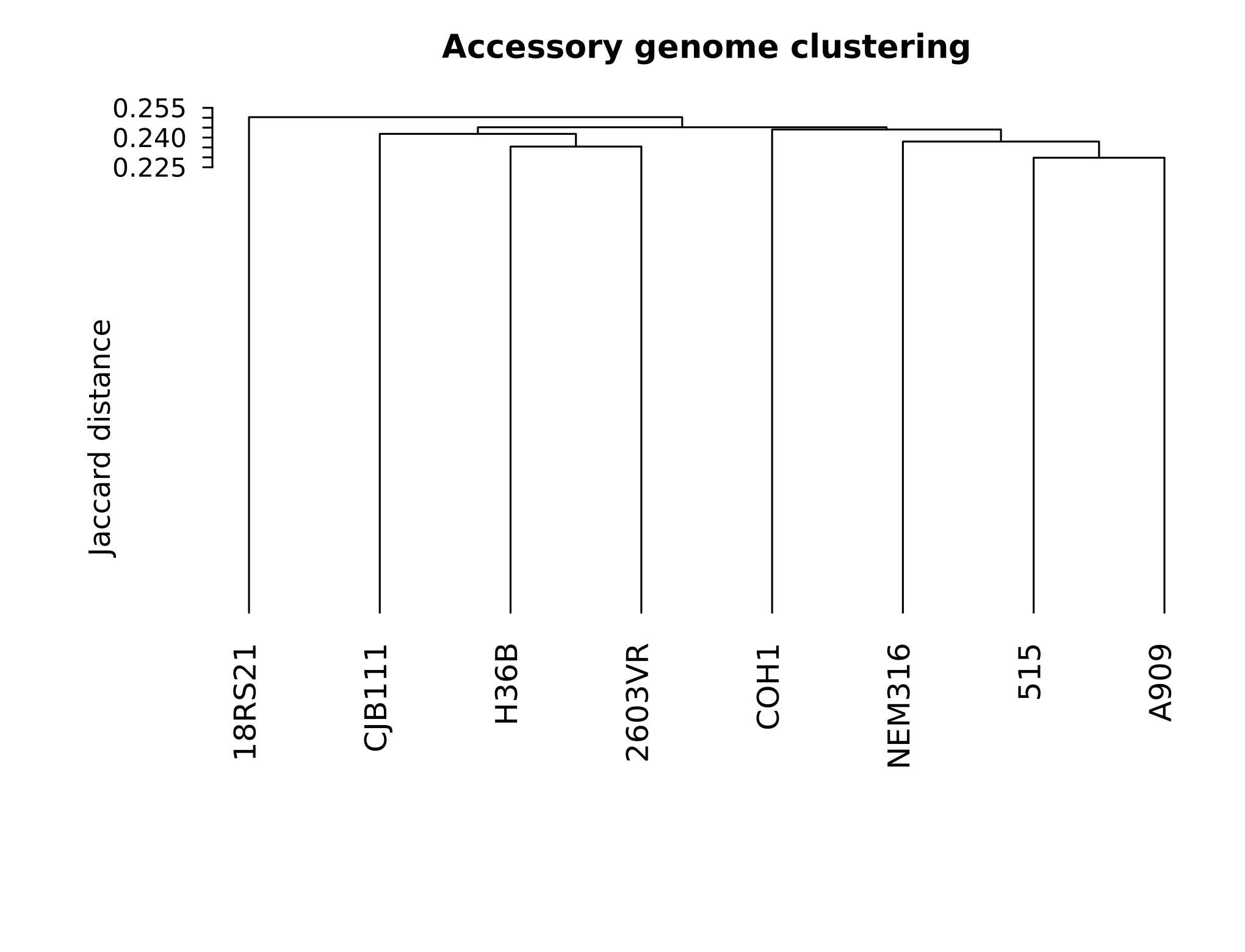
- 计算 gene presence/absence 的 Jaccard 距离。
- 用平均连接法构建附属基因组聚类树。
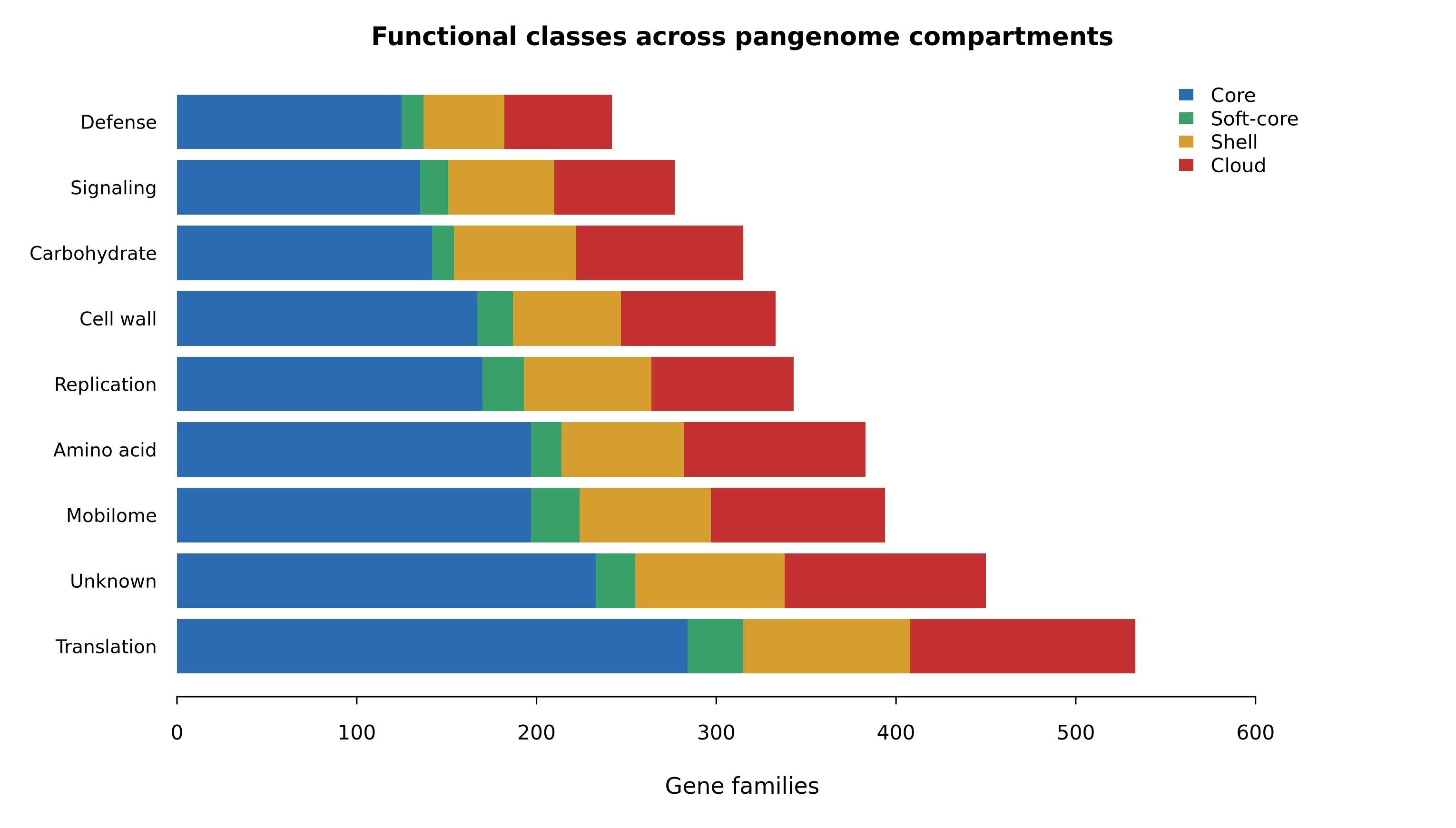
- 汇总功能类别在不同泛基因组组分中的分布。
输出表格:
results/tables/pangenome_class_summary.tsv
results/tables/per_genome_gene_family_counts.tsv
results/tables/pan_core_accumulation_curves.tsv
results/tables/gene_prevalence_distribution.tsv
results/tables/jaccard_distance_matrix.tsv
results/tables/functional_class_by_pangenome_class.tsv
results/tables/accessory_gene_jaccard_tree.nwk
9. 图件索引与图注
所有图件均同时输出 jpg 和 pdf:
| 图号 | 文件 | 源数据 | 绘图脚本 | 图意 |
|---|---|---|---|---|
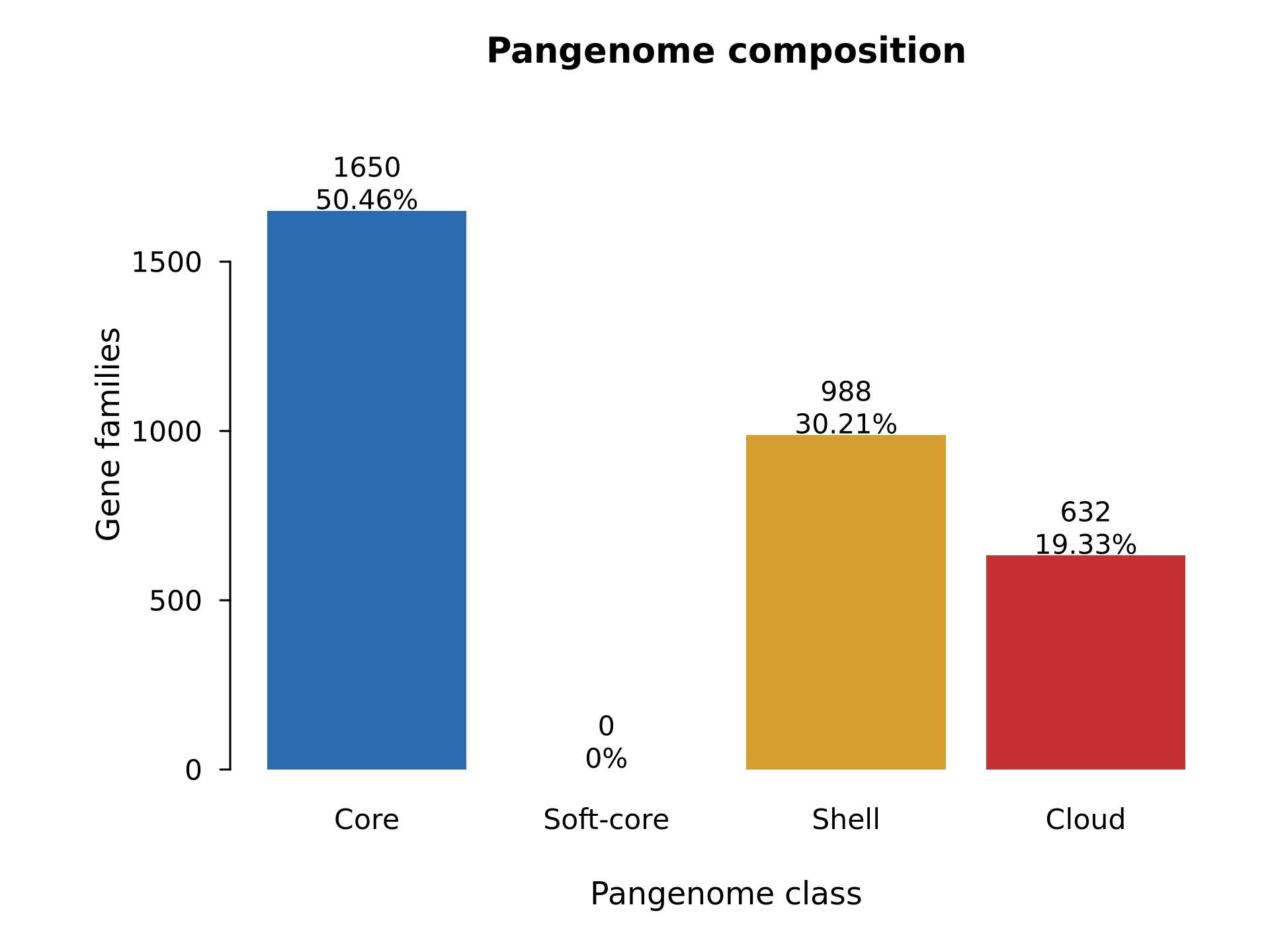
| Figure 1 | results/figures/Figure1_pangenome_composition.jpg/.pdf |
pangenome_class_summary.tsv |
R/03_plot_figures.R |
核心、软核心、壳层和云基因数量 |
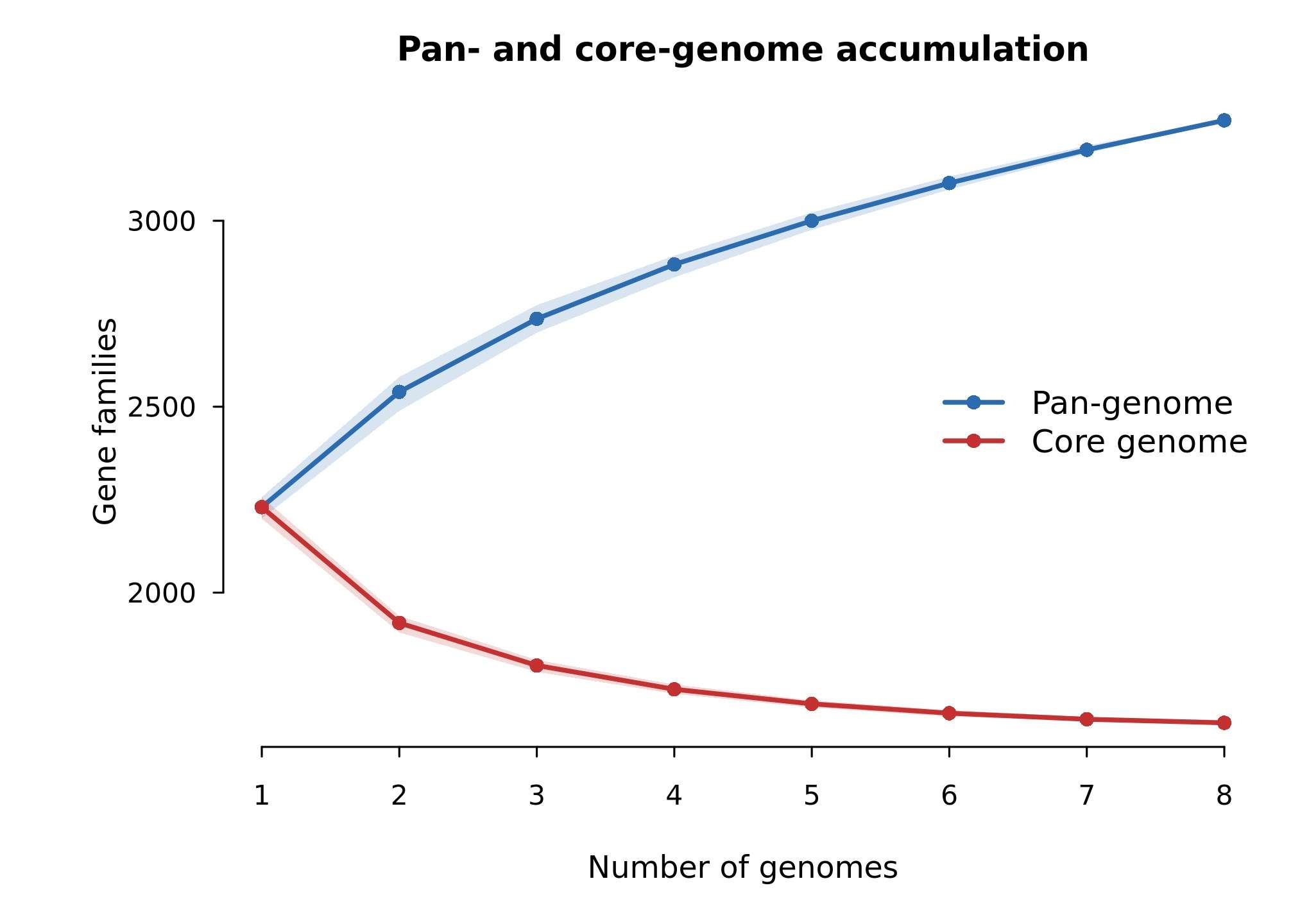
| Figure 2 | results/figures/Figure2_pan_core_accumulation.jpg/.pdf |
pan_core_accumulation_curves.tsv |
R/03_plot_figures.R |
pan-genome 和 core-genome 随基因组数量变化 |
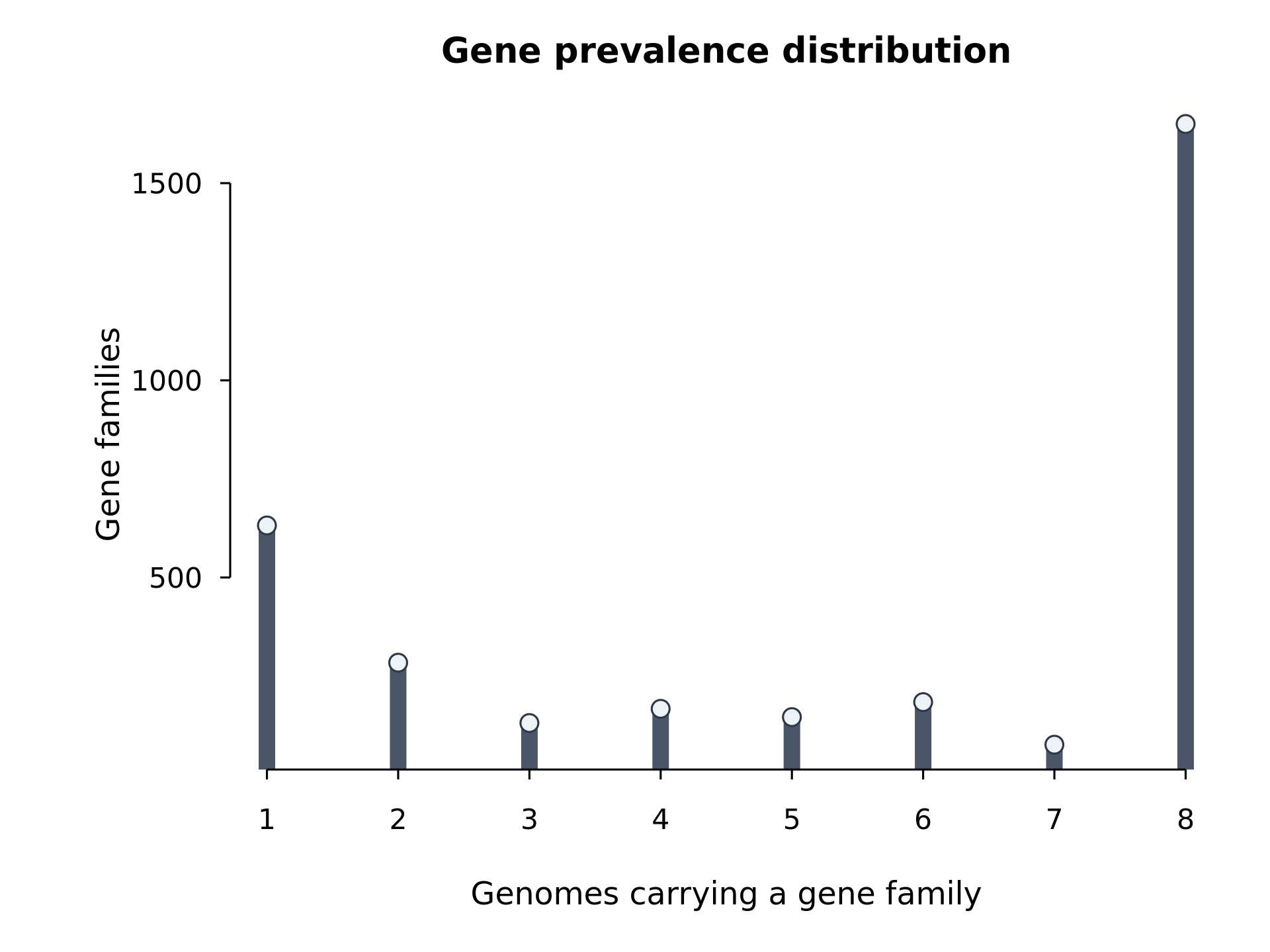
| Figure 3 | results/figures/Figure3_gene_prevalence_distribution.jpg/.pdf |
gene_prevalence_distribution.tsv |
R/03_plot_figures.R |
gene family 在样本中的流行度分布 |
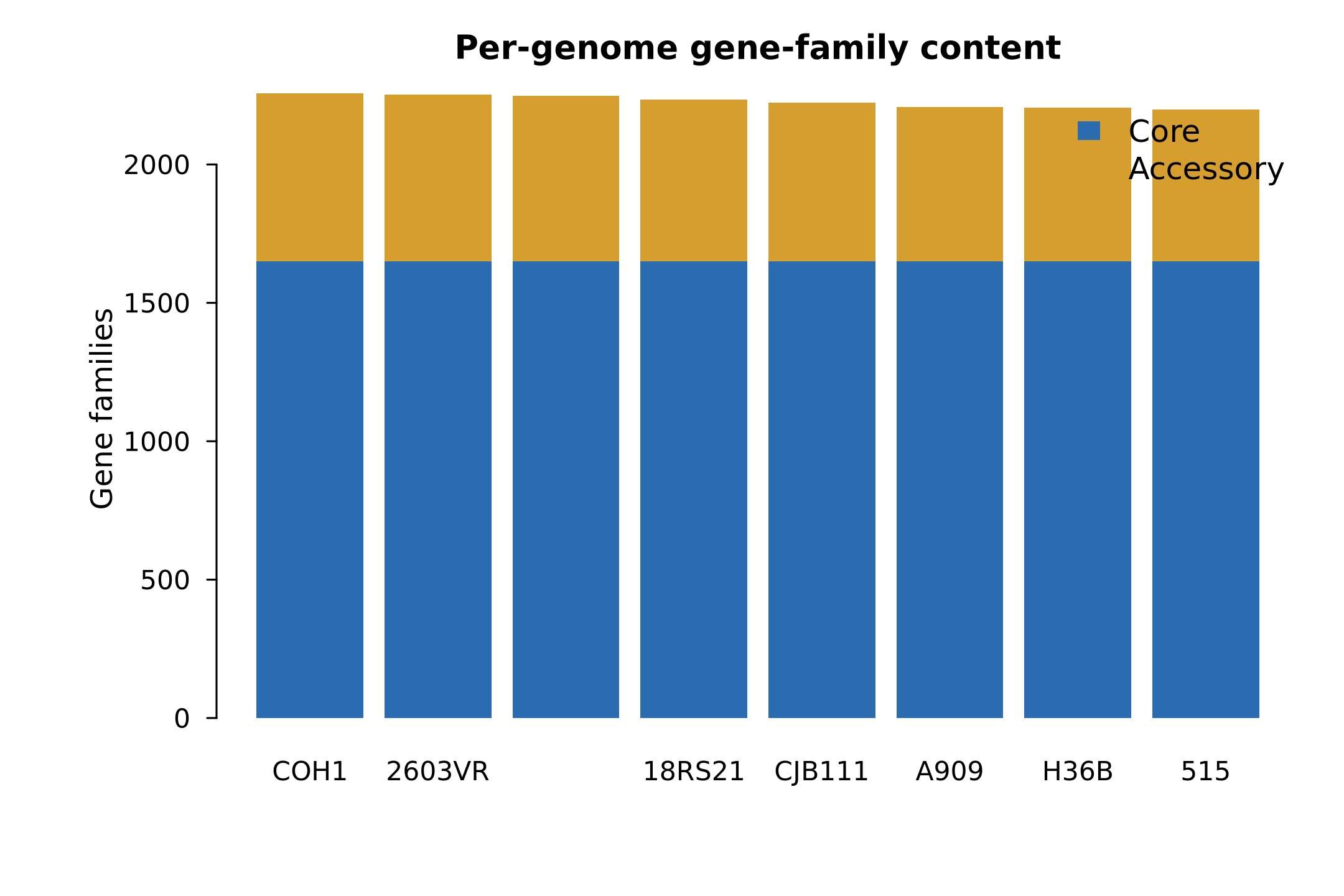
| Figure 4 | results/figures/Figure4_per_genome_gene_content.jpg/.pdf |
per_genome_gene_family_counts.tsv |
R/03_plot_figures.R |
单个基因组的核心和附属基因数量 |
| Figure 5 | results/figures/Figure5_accessory_jaccard_clustering.jpg/.pdf |
jaccard_distance_matrix.tsv |
R/03_plot_figures.R |
附属基因组 Jaccard 距离聚类 |
| Figure 6 | results/figures/Figure6_functional_class_distribution.jpg/.pdf |
functional_class_by_pangenome_class.tsv |
R/03_plot_figures.R |
功能类别在不同泛基因组组分中的分布 |
示例图注可直接作为报告模板:
若要获得本期教程数据和代码,在后台回复:20260624
10. 参考文献
- Tettelin H, Masignani V, Cieslewicz MJ, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial pan-genome. PNAS, 2005. PMID: 16172379. https://pubmed.ncbi.nlm.nih.gov/16172379/
- PMC 开放全文和数据沉积说明。https://pmc.ncbi.nlm.nih.gov/articles/PMC1216834/
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics, 2014. https://pubmed.ncbi.nlm.nih.gov/24642063/
- Page AJ, Cummins CA, Hunt M, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics, 2015. https://pubmed.ncbi.nlm.nih.gov/26198102/
- Tonkin-Hill G, MacAlasdair N, Ruis C, et al. Producing polished prokaryotic pangenomes with Panaroo. Genome Biology, 2020. https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02090-4
- Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 2018. https://pubmed.ncbi.nlm.nih.gov/30423086/
- Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm. Journal of Computational Biology, 2012. https://pubmed.ncbi.nlm.nih.gov/22506599/
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics, 2013. https://pubmed.ncbi.nlm.nih.gov/23422339/
- Walker BJ, Abeel T, Shea T, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE, 2014. https://pubmed.ncbi.nlm.nih.gov/25409509/
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Research, 2015. https://pubmed.ncbi.nlm.nih.gov/25977477/
- Schwengers O, Jelonek L, Dieckmann MA, et al. Bakta: rapid and standardized annotation of bacterial genomes. Microbial Genomics, 2021. https://doi.org/10.1099/mgen.0.000685
若要获得本期教程数据和代码,在后台回复:20260624
若我们的教程对你有所帮助,请
点赞+收藏+转发,这是对我们最大的支持。
小杜的生信筆記 ,注于分享生物信息学相关知识和R语言绘图教程。
发表论文投稿及招聘信息请使用word格式或MarkDown格式发送到
bioinfor2025@163.com
,均为无偿。
2025已离你我而去,2026加油!!
往期部分文章
1. 最全WGCNA教程(替换数据即可出全部结果与图形)
推荐大家购买最新的教程,若是已经购买以前WGNCA教程的同学,可以在对应教程留言,即可获得最新的教程。(注:此教程也仅基于自己理解,不仅局限于此,难免有不恰当地方,请结合自己需求,进行改动。)
2. 精美图形绘制教程
3. 转录组分析教程
4. 转录组下游分析
小杜的生信筆記 ,注于分享生物信息学相关知识和R语言绘图教程。

汇聚全球AI编程工具,助力开发者即刻编程。
更多推荐


 2
2 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)